Difference between revisions of "QMCChem"
Jump to navigation
Jump to search
| Line 26: | Line 26: | ||
* [{{SERVER}}/qmcchem_doc/QMC_Chem_Documentation.html Documentation] | * [{{SERVER}}/qmcchem_doc/QMC_Chem_Documentation.html Documentation] | ||
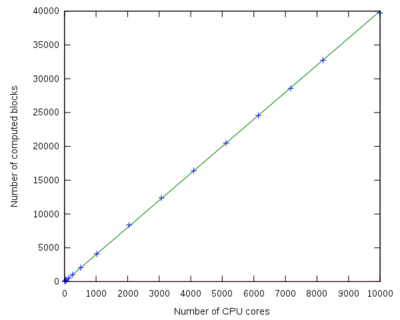
| − | + | == Parallel speed-up curve == | |
[[File:qmcchem_speedup.png|400px|left]] | [[File:qmcchem_speedup.png|400px|left]] | ||
Revision as of 00:59, 10 January 2011
One of our main activities is the development of the massively parallel QMC code QMC=Chem.
Contents
Current Features
Methods
- VMC
- DMC
- Jastrow factor optimization
- CI coefficients optimization
Wave functions
- Single determinant
- Multi-determinant
- Multi-Jastrow
- Nuclear cusp correction
Properties
Practical aspects
- Very low memory requirements
- Runs on a large number of processors : tested on 512 processors at the CALMIP cluster, and on 1000 processors on the EGEE European grid.
- Checkpointing
- Fail safe : if the code is aborted, the data is kept. This feature is very useful in grid environments.
- Easy development with the IRPF90 tool.
- Documentation
Parallel speed-up curve

| Number of processors | Number of computed blocks | Speed-up |
|---|---|---|
| 1 | 5 | 1 |
| 8 | 41 | 8 |
| 16 | 81 | 16 |
| 32 | 170 | 32 |
| 64 | 321 | 64 |
| 128 | 644 | 128 |
| 256 | 1280 | 256 |
| 512 | 2417 | 483 |
Features under development
Properties
- Molecular Forces
- Moments (dipole, quadrupole,...)
- Electron density
- ZV-ZB EPLF estimator
Practical aspects
- Graphical interface for input and output
Input file creation
The QMC=Chem input file can be created using the web interface. Upload a Q5Cost file or an output file from GAMESS, Gaussian or Molpro, and you will download the QMC=Chem input directory.